
At the time the Commission published its “Proposal for a regulation to simplify rules on medical and in vitro diagnostic devices”, making amendments to Regulations (EU) 2017/745 (“EU MDR”) and (EU) 2017/746 (the “Proposal”), it noted that there would be changes to the classification rules. There is a lot of talk that the proposed amendments to Rule 11 (the “Proposed Amendments”) will, if adopted, result in lower risk classes for certain medical devices, including medical device software (“MDSW”). Is this really the case? We review the Proposed Amendments in detail below.
Why was Rule 11 originally introduced?
Under the EU Medical Device Directive 93/42/EEC (the “MDD”) most standalone software ended up a “low risk” class I device under the “catch all” Rule 12[1] of the MDD. This was the case, even if the software could have potentially significant impacts on health outcomes.
The Commission realised this issue and stated in a “Communication” in 2017 it needed to update the classification rules “to keep pace with technological and scientific progress.” As part of its classification overhaul, the Commission introduced MDSW-specific Rule 11 into the EU MDR into section 6.3, Chapt. III of Annex VIII.
How are devices classified under the current Rule 11?
The interpretation of classification of software under Rule 11 is set out in guidance document MDCG 2019-11. This guidance explains that Rule 11 can be split into three sub-rules:
Rule 11a states:
“Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
— death or an irreversible deterioration of a person's state of health, in which case it is in class III; or
— a serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb.”
MDCG 2019-11 states that Rule 11a is generally applicable to most MDSW. It also states it “mirrors” the classification guidance in the International Medical Device Regulatory Forum’s (“IMDRF”) “Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations” (“IMDRF Framework”). Like the IMDRF Framework, someone classifying a device under Rule 11a (as interpreted by MDCG 2019-11) has to consider both the: (i) state of health/condition of the patient; and (ii) significance of the information provided by the software to the healthcare decision.
However, Rule 11a does not fully mirror the IMDRF Framework. The IMDRF Framework covers a broader scope of products (e.g., products intended just for monitoring would fall within the scope). Additionally, the IMDRF Framework provides for certain devices used in non-serious situations and/or providing low significance information to be the lowest risk classification. Whereas, the lowest risk classification for devices falling within Rule 11a is class IIa.
This is shown in Figure 1 and illustrates how the risk class under Rule 11a is applied to different products. Under the IMDRF Framework the products that would be classified according to the rules highlighted by red squares would be class I, rather than class IIa.
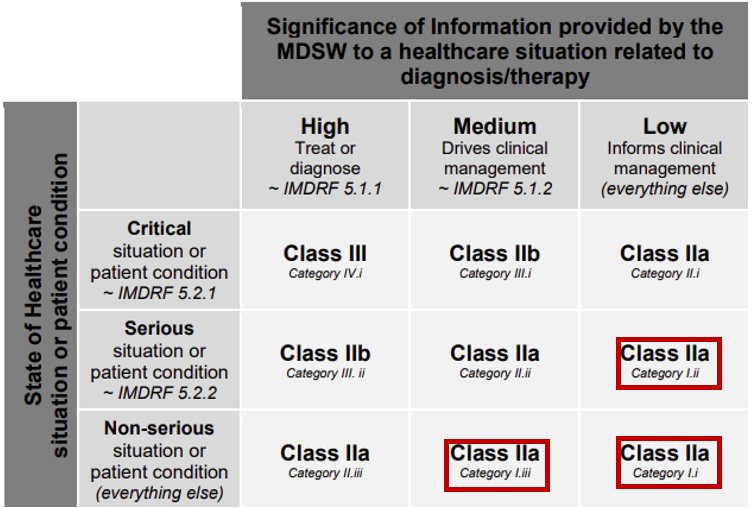
Figure 1 – Annex III of MDCG 2019-11
Rule 11b states:
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
This rule was introduced to cover devices only used for monitoring (as Rule 11a only covered devices for “diagnostic and therapeutic purposes”). It mirrors Rule 10 that covered hardware intended to monitor physiological processes. As noted above, this Rule does not align with the IMDRF Framework.
Finally, Rule 11c is a “catch all” for anything not covered by Rules 11a and 11b: “All other software is classified as class I.”
As Rule 11a is so broadly drafted and Rule 11b covers all monitoring devices, very few devices fall under Rule 11c. As such, almost no devices are class I under the current Rule 11 of the EU MDR. Limited examples are found in Annex IV of MDCG 2019-11 and include (a) software intended to support conception; and (b) an app intended to assist persons with a communication disorder.
A practical implication of Rule 11 as it is currently drafted is that the vast majority of both new and existing MDSW have been up classified to class IIa or above. Under the MDD, most MDSW were in class I and so self-certified by the legal manufacturer. This meant that Notified Bodies had relatively little familiarity with MDSW products. As a result, when the EU MDR came into effect Notified Bodies were (and, to an extent, still are) overloaded with MDSW undergoing conformity assessments despite having little familiarity with such products. Additionally, there is arguably over regulation of lower risk devices, which would be class I devices following the IMDRF Framework.
The essential criticism of Rule 11 is that its classification logic focuses on the severity of potential downstream clinical consequences, while largely ignoring the probability of that consequence and the materiality or clinical weight of the information provided by the MDSW. Rule 11 fails to allow for an assessment of the materiality of the information provided by the MDSW. In doing so, Rule 11 places undue emphasis on the severity of a potential outcome of a decision rather than the probability of such an outcome.
What are the Proposed Amendments to Rule 11?
Firstly, the proposed Rule 11 is no longer split into three sub-rules. Instead, the Rule 11a has been adapted to more closely align with the IMDRF Framework and to cover the products that would fall into Rule 11b. Also Rule 11c has been deleted (so any devices falling outside of Rule 11 would be a class I under Rule 13). We have highlighted key changes in different colours to analyse these below:

What are the impacts of the key changes?
1. Scope of the proposed Rule 11:
The Proposed Amendments have removed the wording “intended to provide information which is used to take decisions with diagnosis or therapeutic purposes.” This has been replaced with software that (a) provides a “clinical benefit;” and (b) falls within a list of medical purposes (e.g., diagnosis, treatment etc.). Therefore, a key question is whether this impacts the scope of the devices covered by this rule.
Clinical benefit is defined in Article 2(53) of the EU MDR as “the positive impact of a device on the health of an individual, expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health.” Requiring MDSW to confer a clinical benefit does not impact the scope of Rule 11 as any medical device should have a positive impact on patient health in order to comply with the General Safety and Performance Requirements in Annex I of the EU MDR.
However, by writing a list of specific medical purposes (rather than using the broad language “diagnosis or therapeutic purposes”), it is necessary to compare these to the medical purposes set out in the definition of a medical device (Article 2(2) of the EU MDR). Any MDSW falling within the first bullet point of the Article 2(2) definition is covered[2]. Devices falling into the second bullet point are also covered on the basis that “condition” is interpreted to cover both “injury” and “disability”[3]. Notably, the list now expressly includes “monitoring,” so devices that previously fell under Rule 11b will now be covered by the amended Rule 11 (which aligns with the IMDRF risk classification framework).
However, there is a question whether devices within the third bullet point of Article 2(2) (i.e., software intended for the purpose of “investigation… of the anatomy or of a physiological or pathological process or state”) would be covered. “Investigation” is not defined in the EU MDR but it is referenced in the definition of “diagnosis” in MDCG 2022-05. This states that “‘[d]iagnosis’ is the process of investigation of the anatomy, morphology, the condition or the functions of the human body irrespective if these are physiological or pathological, and subsequent interpretation of this information with a view to determining possible abnormalities. In this context investigation can include visualisation, detection or measurement” (emphasis added).
Under this definition, “investigation” is part of diagnosis. However, in practice, there are virtually no MDSW intended for “investigation” that would not also fall within bullet point 1 or 2 of Article 2(2) of the EU MDR. Therefore, such MDSW for “investigation” would fall within the existing Rule 11 to the extent that it falls within the scope of the intended use of “diagnosis […] of disease.” Such MDSW for “investigation” would also fall within the proposed Rule 11 (rather than being a Class I under the “catch all” in Rule 13) on the same basis[4]. However, MDSW for the “replacement or modification of the anatomy or of a physiological or pathological process or state” would fall outside of the proposed Rule 11, and therefore within Rule 13, as long as they are not also intended for a purpose captured by the first (or second) bullet point of Article 2(2) of the EU MDR.
2. Possibility or probability of causing an impact
The existing Rule 11 classifies MDSW on the basis of the impact their decisions “may cause.” This means that if there is any possibility (however remote) that a MDSW has a particular impact, it will have the applicable classification. For example, if there is any chance the software could cause death or an irreversible deterioration, it would be class III.
The Proposed Amendments to Rule 11 remove this language and instead refer to software “with a risk of causing” a particular impact. Risk is not defined in the EU MDR, but the concept of “risk” includes both the likelihood/probability of an event happening and the consequence/severity of that impact.
Whether this will actually make a difference in how MDSW is classified is unclear. Arguably even with a very low likelihood of something happening there could still be a “risk” it occurs. Therefore, we hope the Commission publishes guidance to explain the impact of this change.
3. Change in classification
As a starting position, the Proposed Amendments make the default classification for MDSW Class I, rather than Class IIa.
However, do the Proposed Amendments to Rule 11 down classify any MDSW to class 1? We think not.
The Proposed Amendments to Rule 11 mirror the “state of healthcare situation or condition” of the patient referred to in the IMDRF Framework. While the Proposed Amendment does not separate out the “significance of the information provided by the SaMD to the healthcare decision” in the same way as the IMDRF Framework, it is reasonable to assume the Commission intends the amended Rule 11 to continue to be interpreted in this way (as per the MDCG 2019-11 guidance).
Under the Proposed Amendments, all categories of MDSW within scope of Rule 11 will remain in the same category as in the current Rule 11. This is illustrated in Figure 2 below, which shows how devices would now be classified under the Proposed Amendments.
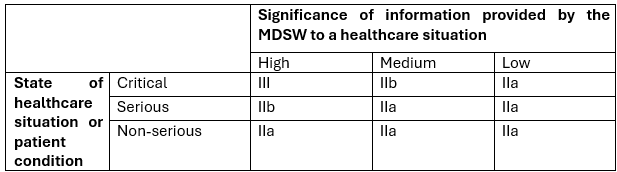
Figure 2 – Bristow’s representation of the Commission’s Proposed Amendments for Rule 11.
Overall Impact
In short, the Proposed Amendment to Rule 11 should not radically change the scope of the devices covered by this Rule.
The Proposed Amendments are also a missed opportunity. As mentioned above, its classification logic emphasises what could happen after a downstream decision rather than how likely it is to happen. Classification rules are supposed to reflect risk, which in turn is a combination of severity and probability. The absence of probability in the classification rule results in an overly conservative classification, and this central issue is not resolved by the Proposed Amendments.
[1] According to Rule 12 “[a]ll other active devices are class I.”
[2] First bullet point of Article 2(2): “diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease”
[3] Second bullet point of Article 2(2): “diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability”
[4] Rule 13 of the EU MDR provides “all other active devices are classified as class I"
/Passle/5f3d6e345354880e28b1fb63/MediaLibrary/Images/2025-09-29-13-48-10-128-68da8e1af6347a2c4b96de4e.png)
/Passle/5f3d6e345354880e28b1fb63/MediaLibrary/Images/2026-03-11-10-54-57-673-69b14a01946e66a5b3c15a3e.jpg)
/Passle/5f3d6e345354880e28b1fb63/MediaLibrary/Images/2024-08-23-11-31-07-354-66c872fb971eecc249d83d40.png)
/Passle/5f3d6e345354880e28b1fb63/SearchServiceImages/2026-07-30-11-35-40-160-6a6b370c7f6e92629d6bd205.jpg)