

The European Commission has recently published a draft implementing regulation under the EU Medical Devices Regulations (“EU MDR”)[1] and In Vitro Diagnostic Medical Devices Regulations (“IVDR”)[2] which will introduce more prescriptive rules for Notified Bodies conducting conformity assessments of medical devices and in vitro diagnostics (“IVDs”). The implementing regulation is out for consultation until 9 January 2026.
The implementing regulation is designed to address three perceived problems which commonly arise in interactions between medical device and IVD manufacturers and Notified Bodies:
- Divergent approaches by some Notified Bodies quoting for conformity assessment activities and onboarding new customers, leading to unexpectedly high costs or even rejected applications.
- Divergent approaches by some Notified Bodies setting and agreeing timelines for completion of conformity assessment and re-certification activities, leading to unexpected delays.
- Some Notified Bodies adopting an unnecessarily intensive approach to re-certification of existing medical devices, leading to duplication of the original conformity assessment review and excessive costs and timelines.
Overall the implementing regulation is intended to standardise some of the business practices of the various Notified Bodies appointed under the EU MDR and IVDR, leading to more consistent manufacturer experiences when dealing with Notified Bodies. The Commission hopes that this will accelerate conformity assessments for new medical devices and IVDs coming to market, helping improve European access to novel medical technologies after years of tumult and uncertainty created by the introduction of EU MDR and IVDR.
Below we summarise some of the issues addressed by the draft implementing regulation.
On Notified Bodies’ quoting and onboarding
The European Commission identifies two problems with Notified Bodies’ current approach to onboarding new customers and providing quotes for the work they will do for them:
- Notified Bodies do not do enough to verify the type of customer they are dealing with and the type of device being submitted for conformity assessment, leading to conformity assessment applications being unexpectedly rejected because the Notified Body is not authorised to assess that device category, or to inappropriate fees being quoted because the Notified Body has not established that the customer is a small and medium-sized enterprise (“SME”).
- Notified Bodies do not provide reliable estimates to manufacturers of the fees that will be charged. For instance, some Notified Bodies fail to provide a breakdown of the costs which will be incurred for separate phases of work, such as technical documentation review, quality management system (“QMS”) audit and market surveillance activities, or fail to specify that travel and accommodation expenses will be charged in addition to fees for on-site audits.
As a result, the implementing regulation will prescribe minimum information that a Notified Body must gather from a prospective customer before issuing a quote and will specify the cost elements that a Notified Body must include in its quote. It will also require a Notified Body to inform the manufacturer in advance of any planned increase in costs for which it previously provided a quote and provide a justification for such an increase.
On timelines for completing conformity assessment activities
The European Commission observes that Notified Bodies have adopted highly variable approaches to agreeing conformity assessment timelines with manufacturers, leading to widely different timelines for comparable conformity assessment procedures without evident justification. The Commission takes the view that conformity assessments should be completed in the shortest feasible timeframe to ensure safe and continuous supply of medical devices and IVDs in the EU.
As a result, the implementing regulation will require Notified Bodies to introduce documented procedures defining how it determines timeframes for completion of conformity assessment activities. The Notified Body will be required to specify the timeframe it has agreed with the manufacturer in the quotation.
In addition, the implementing regulation will prescribe maximum timeframes for different phases of conformity assessment activity:
| Phase | Initiation | Completion | Maximum duration |
| |||
| Receipt by the Notified Body of a completed application. | Signature of contract between Notified Body and manufacturer. | 30 days |
| Initiation of the first audit programme activity by the Notified Body. | Completion of final review referenced in Section 4.7, Annex VII EU MDR/IVDR. | 120 days |
| Initiation of the technical documentation review by the Notified Body. | Completion of final review referenced in Section 4.7, Annex VII EU MDR/IVDR. | 90 days |
| The day after completion of the last relevant final review in relation to the QMS audit or product verification. | Issue of certificate in accordance with Section 4.8, Annex VII EU MDR/IVDR. | 15 days |
| |||
| Receipt by the Notified Body of completed documentation in relation to the proposed change. | The day on which the Notified Body notifies the manufacturer of its decision whether additional conformity assessment activities are needed or of the approval of the planned change. | 30 days |
| Initiation of the first audit programme activity or technical documentation review by the Notified Body, whichever is earlier. | The day on which the Notified Body notifies the manufacturer of the approval of the planned change. | 90 days |
| The day after the Notified Body notifies the manufacturer that the planned change is approved, following steps (a) or (b). | Issue of supplement to the relevant certificate | 15 days |
It is notable that not all of these phases of conformity assessment activity are sequential (in that completion of one does not necessarily trigger the initiation of the next). The European Commission will not therefore impose an overall time limit for the completion of all conformity assessment activities from the point at which a completed application is submitted.
The draft implementing regulation will also allow for these maximum timeframes to be interrupted to allow the manufacturer to address requests from the Notified Body, such as requests for further information, and to allow for necessary input from the EMA, regulatory authorities, an expert panel or an EU reference laboratory.
Interruptions to allow manufacturers to respond to Notified Body requests will be capped at a certain number per phase of activity. Presumably this is to prevent Notified Bodies from stalling for time by requesting further information unnecessarily. There are no caps on the number of permitted interruptions for input from the EMA and other authorities.
Notably, Notified Bodies will also be required to implement processes in their own quality management systems to track the duration and costs associated with performing conformity assessment activities, including their performance against the maximum timeframes. From 1 January 2028, they will be obliged to draw up and publish an annual report detailing the findings of these processes. This will be a useful transparency measure for manufacturers, allowing them to shop around for the Notified Body that offers the best performance in terms of cost and timeliness.
On re-certification activities
The European Commission considers that Notified Bodies have adopted highly variable approaches to conducting re-certification activities under the EU MDR and IVDR, leading to overly intensive reviews in some cases and high variability in terms of cost and timeframes to obtain re-certification.
To tackle this problem, the implementing regulation will introduce a more rigorously defined process for conducting re-certification activities. In particular, Notified Bodies will be obliged to implement procedures under which they notify manufacturers one year prior to the expiry of a QMS or device certificate and require the submission of an application for re-certification accompanied by a specific set of supporting documentation. Notified Bodies will be obliged to review the application submitted within 60 days of receipt and to verify specific matters which are relevant to the decision whether to grant the re-certification. Critically, the Notified Body will be obliged to limit its review to the prescribed set of supporting documentation. The Notified Body will not be entitled to request additional information from the manufacturer unless the supporting documentation is insufficient and additional information is necessary to complete the assessment.
Once the re-certification review is complete and a positive decision on recertification issued, the Notified Body will be obliged to re-issue the relevant certificate(s) within 15 days.
Some observations
In principle, the new requirements for Notified Bodies set out in the draft implementing regulation are a good thing. They should go some way to improving the efficiency and accountability of Notified Bodies and to increasing the predictability of the conformity assessment experience for manufacturers.
However, perhaps inevitably, these new rules will be subject to fairly lengthy transitional provisions, so it will take some time for manufacturers to feel the benefit of the changes they introduce. The rules on maximum timeframes will not apply to conformity assessment activities for which the Notified Body and manufacturer signed a written agreement before 3 months after the entry into force of the implementing regulation. The rules on re-certification of existing certificates will not apply to re-certification reviews of certificates expiring before 18 months after the entry into force of the implementing regulation.
Meanwhile, Notified Bodies will not be obliged to track their performance against cost and timeliness metrics until 12 months after the entry into force of the implementing regulation, and the obligation to publish an annual report on these metrics will not come into force until 1 January 2028.
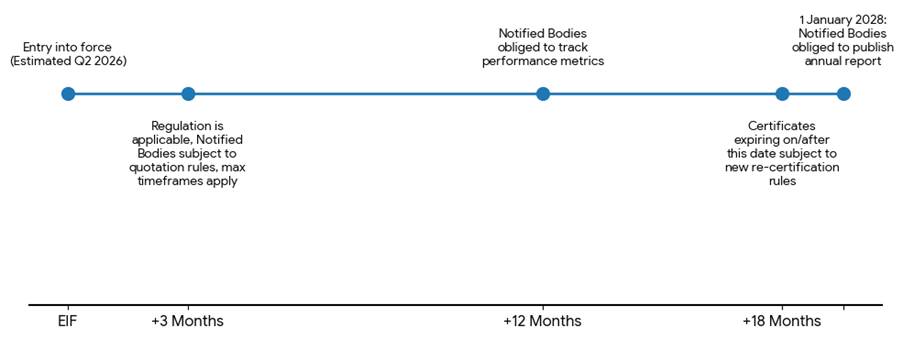
Fig.1[4]
Companies and individuals responding to the consultation have already raised a wide variety of concerns regarding the text: from the potential burden for manufacturers associated with the information that Notified Bodies will be obliged to solicit from them when providing a quote, to the risk of Notified Bodies responding defensively to measures such as maximum timelines for review and transparency around cost and timeliness by deprioritising novel, complex or SME-originating applications which are more likely to lead to cost and timeframe overruns.
[1] Regulation (EU) 2017/745
[2] Regulation (EU) 2017/746
[3] The implementing regulation requires that the QMS audit should run concurrently with the product verification process, subject to the need for technical documentation review output to influence the QMS audit programme.
[4] Generated using AI.
/Passle/5f3d6e345354880e28b1fb63/MediaLibrary/Images/2025-09-29-13-48-10-128-68da8e1af6347a2c4b96de4e.png)
/Passle/5f3d6e345354880e28b1fb63/SearchServiceImages/2026-07-30-11-35-40-160-6a6b370c7f6e92629d6bd205.jpg)
/Passle/5f3d6e345354880e28b1fb63/SearchServiceImages/2026-07-30-15-10-36-161-6a6b696cf10dbec010070cf3.jpg)
/Passle/5f3d6e345354880e28b1fb63/SearchServiceImages/2026-07-30-09-58-02-499-6a6b202a084549892d338aab.jpg)